Difference between revisions of "Kmill104 Week 10"
(→Data & Files: fixing formatting) |
(→Data & Files: adding txt file) |
||
| Line 170: | Line 170: | ||
[[Media:KM_GRNmap.xlsx | Excel Workbook for GRN KM]] | [[Media:KM_GRNmap.xlsx | Excel Workbook for GRN KM]] | ||
| + | |||
| + | [[Media:KM_wt_STEM.txt]] | ||
==Conclusion== | ==Conclusion== | ||
Revision as of 20:37, 3 April 2024
Purpose
Methods/Results
- Prepare your microarray data file for loading into STEM.
- Insert a new worksheet into your Excel workbook, and name it "(STRAIN)_stem".
- Select all of the data from your "(STRAIN)_ANOVA" worksheet and Paste special > paste values into your "(STRAIN)_stem" worksheet.
- Your leftmost column should have the column header "Master_Index". Rename this column to "SPOT". Column B should be named "ID". Rename this column to "Gene Symbol". Delete the column named "Standard_Name".
- Filter the data on the B-H corrected p value to be > 0.05 (that's greater than in this case).
- Once the data has been filtered, select all of the rows (except for your header row) and delete the rows by right-clicking and choosing "Delete Row" from the context menu. Undo the filter. This ensures that we will cluster only the genes with a "significant" change in expression and not the noise.
- Delete all of the data columns EXCEPT for the Average Log Fold change columns for each timepoint (for example, wt_AvgLogFC_t15, etc.).
- Rename the data columns with just the time and units (for example, 15m, 30m, etc.).
- If you had #DIV/0! errors remaining, it will cause problems. To remove these, open the Find/Replace dialog. Search for #DIV/0!, but don't put anything in the replace field. Click "Replace all" to remove the #DIV/0! errors.
- Save your work. Then use Save As to save this spreadsheet as Text (Tab-delimited) (*.txt). Click OK to the warnings and close your file.
- Note that you should turn on the file extensions if you have not already done so.
- Now download and extract the STEM software. Click here to go to the STEM web site.
- Click on the download link and download the
stem.zipfile to your Desktop. - Extract the file by right-clicking on it and selecting "Extract all" from the menu.
- This will create a folder called
stem.- You now need to download the Gene Ontology and yeast GO annotations and place them in this folder.
- Click here to download the file "gene_ontology.obo".
- Click here to download the file "gene_association.sgd.gz".
- Inside the folder, double-click on the
stem.jarto launch the STEM program.
- Click on the download link and download the
- Running STEM
- In section 1 (Expression Data Info) of the the main STEM interface window, click on the Browse... button to navigate to and select your file.
- Click on the radio button No normalization/add 0.
- Check the box next to Spot IDs included in the data file.
- In section 2 (Gene Info) of the main STEM interface window, leave the default selection for the three drop-down menu selections for Gene Annotation Source, Cross Reference Source, and Gene Location Source as "User provided".
- Click the "Browse..." button to the right of the "Gene Annotation File" item. Browse to your "stem" folder and select the file "gene_association.sgd.gz" and click Open.
- In section 3 (Options) of the main STEM interface window, make sure that the Clustering Method says "STEM Clustering Method" and do not change the defaults for Maximum Number of Model Profiles or Maximum Unit Change in Model Profiles between Time Points.
- In section 4 (Execute) click on the yellow Execute button to run STEM.
- If you get an error, there are some known reasons why stem might not work. If you had #DIV/0! errors in your input file, it will cause problems. Re-open your file and open the Find/Replace dialog. Search for #DIV/0!, but don't put anything in the replace field. Click "Replace all" to remove the #DIV/0! errors. Then save your file and try again with stem.
- In section 1 (Expression Data Info) of the the main STEM interface window, click on the Browse... button to navigate to and select your file.
- Viewing and Saving STEM Results
- A new window will open called "All STEM Profiles (1)". Each box corresponds to a model expression profile. Colored profiles have a statistically significant number of genes assigned; they are arranged in order from most to least significant p value. Profiles with the same color belong to the same cluster of profiles. The number in each box is simply an ID number for the profile.
- Click on the button that says "Interface Options...". At the bottom of the Interface Options window that appears below where it says "X-axis scale should be:", click on the radio button that says "Based on real time". Then close the Interface Options window.
- Take a screenshot of this window (on a PC, simultaneously press the
AltandPrintScreenbuttons to save the view in the active window to the clipboard) and paste it into a PowerPoint presentation to save your figures.
- Click on each of the SIGNIFICANT profiles (the colored ones) to open a window showing a more detailed plot containing all of the genes in that profile.
- Take a screenshot of each of the individual profile windows and save the images in your PowerPoint presentation.
- At the bottom of each profile window, there are two yellow buttons "Profile Gene Table" and "Profile GO Table". For each of the profiles, click on the "Profile Gene Table" button to see the list of genes belonging to the profile. In the window that appears, click on the "Save Table" button and save the file to your desktop. Make your filename descriptive of the contents, e.g. "wt_profile#_genelist.txt", where you replace the number symbol with the actual profile number.
- Upload these files to the wiki and link to them on your individual journal page. Note that it will be easier to zip all the files together and upload them as one file. To do this, select all of the files you want to zip together. Then right click and select "Send to" and "Compressed (zipped) folder" from the context menu.
- For each of the significant profiles, click on the "Profile GO Table" to see the list of Gene Ontology terms belonging to the profile. In the window that appears, click on the "Save Table" button and save the file to your desktop. Make your filename descriptive of the contents, e.g. "wt_profile#_GOlist.txt", where you use "wt", "dGLN3", etc. to indicate the dataset and where you replace the number symbol with the actual profile number. At this point you have saved all of the primary data from the STEM software and it's time to interpret the results!
- Upload these files to the wiki and link to them on your individual journal page. Note that it will be easier to zip all the files together and upload them as one file. To do this, select all of the files you want to zip together. Then right click and select "Send to" and "Compressed (zipped) folder" from the context menu.
- A new window will open called "All STEM Profiles (1)". Each box corresponds to a model expression profile. Colored profiles have a statistically significant number of genes assigned; they are arranged in order from most to least significant p value. Profiles with the same color belong to the same cluster of profiles. The number in each box is simply an ID number for the profile.
- Analyzing and Interpreting STEM Results
- Select one of the profiles you saved in the previous step for further intepretation of the data. I suggest that you choose one that has a pattern of up- or down-regulated genes at the cold shock timepoints. Each member of your group should choose a different profile. Answer the following:
- Why did you select this profile? In other words, why was it interesting to you? I chose the 45 profile. This was most interesting to me because it was the most significant profile, and the up-regulation occurs early and then drops.
- How many genes belong to this profile? 549.0
- How many genes were expected to belong to this profile? 47.1
- What is the p value for the enrichment of genes in this profile? 1.00
- Bear in mind that we just finished computing p values to determine whether each individual gene had a significant change in gene expression at each time point. This p value determines whether the number of genes that show this particular expression profile across the time points is significantly more than expected.
- Open the GO list file you saved for this profile in Excel. This list shows all of the Gene Ontology terms that are associated with genes that fit this profile. Select the third row and then choose from the menu Data > Filter > Autofilter. Filter on the "p-value" column to show only GO terms that have a p value of < 0.05. How many GO terms are associated with this profile at p < 0.05? 81
- The GO list also has a column called "Corrected p-value". This correction is needed because the software has performed thousands of significance tests. Filter on the "Corrected p-value" column to show only GO terms that have a corrected p value of < 0.05. How many GO terms are associated with this profile with a corrected p value < 0.05? 11
- Select 6 Gene Ontology terms from your filtered list (either p < 0.05 or corrected p < 0.05).
- cytoplasmic exosome (RNase complex) (GO:0000177)
- nucleic acid binding (GO:0003676)
- ribosomal large subunit export from nucleus (GO:0000055)
- snoRNA binding (GO:0030515)
- mitochondrial translation (GO:0032543)
- nuclear polyadenylation-dependent rRNA catabolic process (GO:0071035)
- Each member of the group will be reporting on his or her own cluster in your research presentation. You should take care to choose terms that are the most significant, but that are also not too redundant. For example, "RNA metabolism" and "RNA biosynthesis" are redundant with each other because they mean almost the same thing.
- Note whether the same GO terms are showing up in multiple clusters.
- Look up the definitions for each of the terms at http://geneontology.org. In your journal entry, will discuss the biological interpretation of these GO terms. In other words, why does the cell react to cold shock by changing the expression of genes associated with these GO terms? Also, what does this have to do with the transcription factor being deleted (for the groups working with deletion strain data)? I'm not completely sure why these specific genes had a change in expression, but I noticed that the genes were concerned with regulating RNA and protein production from mRNA. When cold shock is experienced by the cell, there likely needs to be a change in what mRNA is transcribed and at what rate. Additionally, the formation of proteins and the rate at which they are formed must also be regulated. Second question is not related to the wild type strain
- To easily look up the definitions, go to http://geneontology.org.
- Copy and paste the GO ID (e.g. GO:0044848) into the search field on the left of the page.
- In the results page, click on the button that says "Link to detailed information about <term>, in this case "biological phase"".
- The definition will be on the next results page, e.g. here.
- definition of cytoplasmic exosome (RNase complex) (GO:0000177): A ribonuclease complex that has 3-prime to 5-prime processive hydrolytic exoribonuclease activity producing 5-prime-phosphomonoesters. Participates in a multitude of cellular RNA processing and degradation events preventing nuclear export and/or translation of aberrant RNAs. Restricted to processing linear and circular single-stranded RNAs (ssRNA) only. RNAs with complex secondary structures may have to be unwound or pre-processed by co-factors prior to entering the complex, esp if the 3-prime end is structured.
- nucleic acid binding (GO:0003676): Binding to a nucleic acid.
- ribosomal large subunit export from nucleus (GO:0000055): The directed movement of a ribosomal large subunit from the nucleus into the cytoplasm.
- snoRNA binding (GO:0030515): Binding to a small nucleolar RNA.
- mitochondrial translation (GO:0032543): The chemical reactions and pathways resulting in the formation of a protein in a mitochondrion. This is a ribosome-mediated process in which the information in messenger RNA (mRNA) is used to specify the sequence of amino acids in the protein; the mitochondrion has its own ribosomes and transfer RNAs, and uses a genetic code that differs from the nuclear code.
- nuclear polyadenylation-dependent rRNA catabolic process (GO:0071035) : The chemical reactions and pathways occurring in the nucleus and resulting in the breakdown of a ribosomal RNA (rRNA) molecule, including RNA fragments released as part of processing the primary transcript into multiple mature rRNA species, initiated by the enzymatic addition of a sequence of adenylyl residues (polyadenylation) at the 3' end the target rRNA.
- Select one of the profiles you saved in the previous step for further intepretation of the data. I suggest that you choose one that has a pattern of up- or down-regulated genes at the cold shock timepoints. Each member of your group should choose a different profile. Answer the following:
Stopping point for Thursday's lab. Be sure to upload all of the files requested to your Week 10 individual journal entry by 12:01am on Tuesday, March 26 so that the files can be checked before we do the next step on Tuesday. Also invoke your template on your Week 10 individual journal entry right away.
Using YEASTRACT to Infer which Transcription Factors Regulate a Cluster of Genes (Tuesday, March 26)
In the previous analysis using STEM, we found a number of gene expression profiles (aka clusters) which grouped genes based on similarity of gene expression changes over time. The implication is that these genes share the same expression pattern because they are regulated by the same (or the same set) of transcription factors. We will explore this using the YEASTRACT database.
- Open the gene list in Excel for the one of the significant profiles from your stem analysis that you chose to perform your GO analysis. It should be a cluster with a clear cold shock/recovery up/down or down/up pattern, and should be one of the largest clusters.
- Copy the list of gene IDs onto your clipboard.
- Launch a web browser and go to the YEASTRACT database.
- On the left panel of the window, click on the link to Rank by TF.
- Paste your list of genes from your cluster into the box labeled ORFs/Genes.
- Check the box for Check for all TFs.
- Accept the defaults for the Regulations Filter (Documented, DNA binding or expression evidence)
- Do not apply a filter for "Filter Documented Regulations by environmental condition".
- Rank genes by TF using: The % of genes in the list and in YEASTRACT regulated by each TF.
- Click the Search button.
- Answer the following questions:
- In the results window that appears, the p values colored green are considered "significant", the ones colored yellow are considered "borderline significant" and the ones colored pink are considered "not significant". How many transcription factors are green or "significant"? 33
- Copy the table of results from the web page and paste it into a new Excel workbook to preserve the results.
- Copy by selecting and dragging down on the table.
- When pasting into Excel, remember to Paste special > Paste values.
- Upload the Excel file to the wiki and link to it in your electronic lab notebook.
- Are CIN5 or GLN3 on the list? If so, what is their "% in user set", "% in YEASTRACT", and "p value"?
- CIN5 - % in user set: 30.63%, % in scerevisiae: 7.22%, p-value: 0.522044721
- GLN3 - % in user set: 38.38%, % in scerevisiae: 8.46%, p-value: 0.002604838
Creating and Visualizing Your Gene Regulatory Network with GRNsight (Tuesday, March 26)
For the mathematical model that we will build, we need to define a gene regulatory network of transcription factors that regulate other transcription factors. We can use GRNsight to assist us with creating the network. We want to generate a network with approximately 15-20 connected transcription factors in it.
- You need to select from the list of "significant" transcription factors that you found in YEASTRACT, which ones you will use to run the model. You will use these transcription factors and add GLN3 and CIN5 if they are not in your list.
- Generally, you should include the top transcription factors with the smallest p values. Explain in your electronic notebook how you decided on which transcription factors to include. Record the list and your justification in your electronic lab notebook. Each group member will select a different network (they can have some overlapping transcription factors, but some should also be different).
- GCN4 GAT1 RPN4 YAP1 MET28 RPH1 MET32 MSN2 RIM101 MET31 SNF2 SFP1 ASG1 PDR1 MSN4 UME6 INO4 HAP1 GLN3 CIN5
- I chose these transcription factors because they were in the top 23 of genes with the smallest p-values.
- Generally, you should include the top transcription factors with the smallest p values. Explain in your electronic notebook how you decided on which transcription factors to include. Record the list and your justification in your electronic lab notebook. Each group member will select a different network (they can have some overlapping transcription factors, but some should also be different).
- Go to the GRNsight beta website.
- Under the "Network" panel on the left-hand side, click the button "Load from database".
- Type the standard name of the transcription factor in the "Select gene" field and click the find button (magnifying glass).
- Continue to add transcription factors in this way until you have added 15-20.
- Click the "Generate Network" button.
- Your network should appear on the screen.
- Check to see if all of the rectangular boxes (nodes) are connected by at least one arrow to another node. If there is a node that is not connected, go back to the "Load from database" button and select your transcription factors again, leaving out the node that was disconnected.
- Record in your electronic lab notebook the number of genes and edges in your network (found at the upper right of the menu bar). 20 genes, 37 edges
- Under the "Layout" section, click on the "Grid Layout" button.
- Export the network image by going to the Export menu and selecting "Export Image > To PNG". Upload the file to the wiki and display it on your individual journal page.
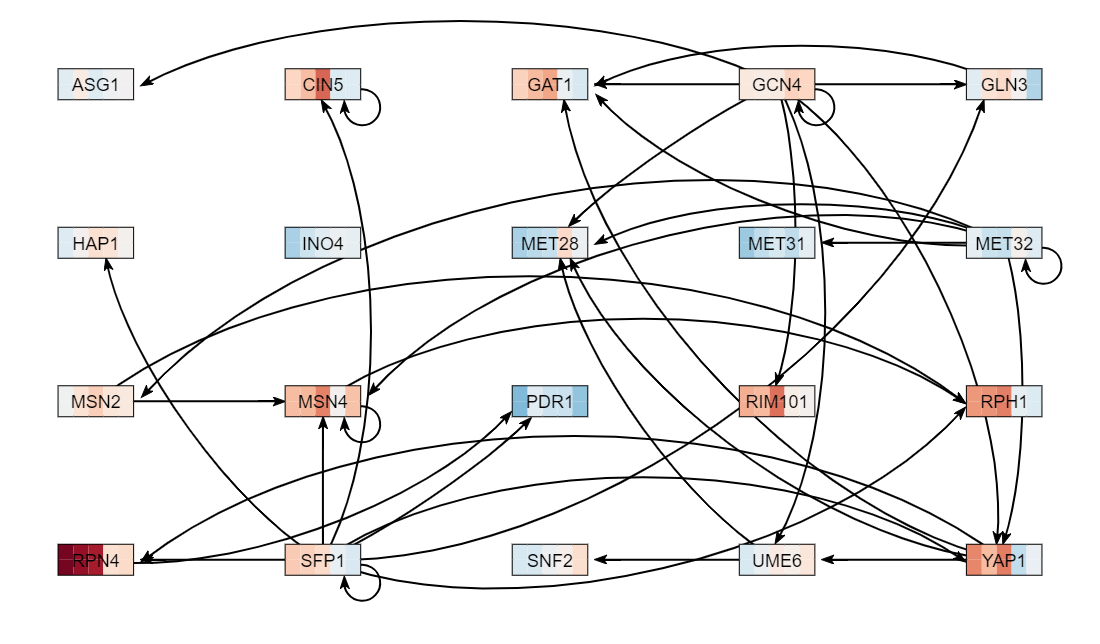
- Export the network image by going to the Export menu and selecting "Export Image > To PNG". Upload the file to the wiki and display it on your individual journal page.

Creating the GRNmap Input Workbook (Tuesday, March 26)
We will also use GRNsight to automatically generate the input workbook for the GRNmap modeling software. Note that this feature is still under development, and we will be performing quality control on the exported workbook.
- With your final network still open in GRNsight, select from the Export menu "Export Data > To Excel". In the window that appears, select the following:
- Under "Select the Expression Data Source:", choose "Dahlquist_2018"
- Under "Select Workbook Sheets to Export:", select the following:
- Network sheets
- "network"
- Expression sheets
- dcin5_log2_expression
- dgln3_log2_expression
- wt_log2_expression
- Additional sheets
- "degradation_rates"
- "optimization_parameters"
- "production_rates"
- "threshold_b"
- Network sheets
- Click the "Export Workbook" button.
- Open your workbook in Excel to perform quality control. Check that it has the following sheets with the following content:
- The "network" sheet should have an adjacency matrix with your selected regulatory transcription factors across the top row and in the first column.
- The "dcin5_log2_expression", "dgln3_log2_expression", and "wt_log2_expression" sheets should have log2 fold changes for each of your selected regulatory transcription factors for each time point (15, 30, 60, 90, 120). Replicate values have the same column headers. If a particular gene is missing all 4-5 replicate values at a particular timepoint for a particular strain, we need to exclude it from the analysis. Go back to generating your network and repeat the steps to generate the network and export to Excel without that gene. Record this in your electronic lab notebook.
- The "production_rates" and "degradation_rates" sheets should have values for each gene.
- The "threshold_b" sheet should have a value of 0 for each gene.
- In the "optimization_parameters" sheet, change the "alpha" value to 0.02 instead of 0.002.
- Insert a new worksheet and name it "network_weights".
- Copy the entire content of the "network" sheet into the "network_weights" sheet.
- Save and upload your Excel Workbook to the wiki and link to it on your individual journal page. Dr. Dahlquist will run your input workbook in the GRNmap modeling software over the break.
Stopping point for Tuesday, March 26. Be sure to complete up to this point for the interim deadline of 11:59pm on Tuesday, April 2 so your files can be checked before class.
Data & Files
Your data and files section should include:
- Your Excel workbook with all of your calculations.
- Your YEASTRACT results should be saved as a new worksheet in this workbook.
- Note that you will be working with this workbook for the next week or two, adding computations to it. Save the new versions to the wiki with the same filename. The wiki will store each version of the file so you can always go back to a previous version, if need be.
- Your PowerPoint slide with a summary table of p values, updated with the screenshots from the stem software.
- You will also be adding to the PowerPoint presentation during subsequent steps in the analysis.
- The input .txt file that you used to run stem.
- The zipped together genelist and GOlist files for each of your significant profiles.
- The .png file showing your gene regulatory network.
- Your exported and quality-checked Excel workbook from GRNsight beta.
Conclusion
Acknowledgements
I worked under the guidance of Dr. Dahlquist on 3-21-24 and 3-26-24. I either consulted Dr. Dahlquist or my classmates if I ever had questions about using STEM, YEASTRACT, or GRNsight. For the procedure section of this individual journal, I first copied the general procedure from the Week 10 assignment page, and then adjusted it so that the specific steps I followed are described above.
Except for what is noted above, this individual journal entry was completed by me and not copied from another source.
Kmill104 (talk) 22:41, 2 April 2024 (PDT)
References
- Dahlquist, K. D., Fitzpatrick, B. G., Camacho, E. T., Entzminger, S. D., & Wanner, N. C. (2015). Parameter estimation for gene regulatory networks from microarray data: cold shock response in Saccharomyces cerevisiae. Bulletin of mathematical biology, 77(8), 1457-1492. DOI: 10.1007/s11538-015-0092-6
- Ernst, J., & Bar-Joseph, Z. (2006). STEM: a tool for the analysis of short time series gene expression data. BMC bioinformatics, 7(1), 191. DOI: 10.1093/bioinformatics/bti1022
- LMU BioDB 2024. (2024). Week 10. Retrieved April 2, 2024, from https://xmlpipedb.cs.lmu.edu/biodb/spring2024/index.php/Week_10
- Teixeira, M. C., Viana, R., Palma, M., Oliveira, J., Galocha, M., Mota, M. N., ... & Monteiro, P. T. (2023). YEASTRACT+: a portal for the exploitation of global transcription regulation and metabolic model data in yeast biotechnology and pathogenesis. Nucleic Acids Research, 51(D1), D785-D791. DOI: 10.1093/nar/gkac1041
User Page
Assignment Pages
- Week 1
- Week 2
- Week 3
- Week 4
- Week 5
- Week 6
- Week 8
- Week 9
- Week 10
- Week 11
- Week 12
- Week 13
- Week 14
- Week 15
Individual Journal Entry Pages
- Kmill104 Week 1
- Kmill104 Week 2
- MSymond1 KMill104 Week 3
- Monarch Initiative Week 4
- Kmill104 Week 5
- Kmill104 Week 6
- Kmill104 Week 8
- Kmill104 Week 9
- Kmill104 Week 10
- Kmill104 Week 11
- Kmill104 Week 12
- Data Analysts Week 13
- Data Analysts Week 14
- Data Analysts Week 15