Difference between revisions of "Kmill104 Week 10"
(→Creating and Visualizing Your Gene Regulatory Network with GRNsight (Tuesday, March 26): updating network details) |
(→Acknowledgements: adding updated talk signature) |
||
| (10 intermediate revisions by the same user not shown) | |||
| Line 1: | Line 1: | ||
==Purpose== | ==Purpose== | ||
| + | The purpose of this week's journal assignment was to analyze a DNA microarray dataset through "high level" analysis. This included using the STEM software for clustering, YEASTRACT for finding which transcription factors regulate these gene clusters, and producing a gene regulatory network in GRNsight. We also worked to keep an organized and detail laboratory notebook, so that another person could follow these steps and reproduce the same results. | ||
==Methods/Results== | ==Methods/Results== | ||
| − | # ''' | + | # '''For this this week's journal assignment, my first step was to prepare my microarray data file for loading into STEM.''' |
| − | #* | + | #* I inserted a new worksheet into my Excel workbook, and named it "wt_stem". |
| − | #* | + | #* I then selected all of the data from my "wt_ANOVA" worksheet and Paste special > pasted values into my "wt_stem" worksheet. |
| − | #** | + | #** I then renamed the column with the header "Master_Index" to "SPOT". I also renamed the column with the column header "ID" to "Gene Symbol". I deleted the column with the header "Standard_Name". |
| − | #** | + | #** I filtered the data on the B-H corrected p value column to be > 0.05. |
| − | #*** Once the data | + | #***Once the data had been filtered, I selected all of the rows (except for my header row) and deleted the rows by right-clicking and choosing "Delete Row" from the context menu. I then undid the filter. This step ensured that the genes I will cluster will only the genes that had a "significant" change in expression. |
| − | #** | + | #** I then deleted all of the other data columns '''''EXCEPT''''' for the Average Log Fold change columns for each timepoint. |
| − | #** | + | #** I then renamed the data columns with just the time and units, so for any column with wt_AvgLogFC_t#, the header became #m. |
| − | + | #** I saved my work. Then, I used ''Save As'' to save this spreadsheet as Text (Tab-delimited) (*.txt), clicked OK to the warnings and closed my file. | |
| − | #** | + | # '''Then, I downloaded and extracted the STEM software.''' |
| − | + | #* I clicked on the [http://www.sb.cs.cmu.edu/stem/stem.zip download link] and downloaded the <code>stem.zip</code> file to my Desktop. | |
| − | # ''' | + | #* I extracted the file by right-clicking on it and selecting "Extract all" from the menu. |
| − | #* | + | #* This created a folder called <code>stem</code>. |
| − | #* | + | #** I then downloaded the Gene Ontology and yeast GO annotations, which I placed in this same folder. I used [https://lmu.box.com/s/t8i5s1z1munrcfxzzs7nv7q2edsktxgl "gene_ontology.obo"] to download the file for Gene Ontology. |
| − | #* This | + | #** I used [https://lmu.box.com/s/zlr1s8fjogfssa1wl59d5shyybtm1d49 "gene_association.sgd.gz"] to download the yeast GO annotations. |
| − | #** | + | #*Inside the folder, I double-clicked on the <code>stem.jar</code> to launch the STEM program. |
| − | |||
| − | #** | ||
| − | #*Inside the folder, double- | ||
# '''Running STEM''' | # '''Running STEM''' | ||
| − | ## In section 1 (Expression Data Info) of the the main STEM interface window, | + | ## In section 1 (Expression Data Info) of the the main STEM interface window, I clicked on the ''Browse...'' button to navigate to and select my file. |
| − | ##* | + | ##* I then clicked on the radio button ''No normalization/add 0''. |
##* Check the box next to ''Spot IDs included in the data file''. | ##* Check the box next to ''Spot IDs included in the data file''. | ||
| − | ## In section 2 (Gene Info) of the main STEM interface window, | + | ## In section 2 (Gene Info) of the main STEM interface window, I left the default selection for the three drop-down menu selections for Gene Annotation Source, Cross Reference Source, and Gene Location Source as "User provided". |
| − | ## | + | ## I clicked the "Browse..." button to the right of the "Gene Annotation File" item. I then browsed to my "stem" folder and selected the file "gene_association.sgd.gz" and clicked Open. |
| − | ## In section 3 (Options) of the main STEM interface window, | + | ## In section 3 (Options) of the main STEM interface window, I checked that the Clustering Method says "STEM Clustering Method" and did not change the defaults for Maximum Number of Model Profiles or Maximum Unit Change in Model Profiles between Time Points. |
| − | ## In section 4 (Execute) | + | ## In section 4 (Execute), I clicked on the yellow Execute button to run STEM. |
| − | |||
# '''Viewing and Saving STEM Results''' | # '''Viewing and Saving STEM Results''' | ||
| − | ## A new window | + | ## A new window opened called "All STEM Profiles (1)". Each box corresponded to a model expression profile. Colored profiles had a statistically significant number of genes assigned; they were arranged in order from most to least significant p value. Profiles with the same color belonged to the same cluster of profiles. The number in each box was simply an ID number for the profile. |
| − | ##* | + | ##* I clicked on the button that says "Interface Options...". At the bottom of the Interface Options window that appears below where it says "X-axis scale should be:", I clicked on the radio button that says "Based on real time". I then closed the Interface Options window. |
| − | ##* | + | ##*I took a screenshot of this window, and then pasted it into a PowerPoint presentation that also contained my p-value table. |
| − | ## | + | ## I then clicked on each of the SIGNIFICANT profiles to open a window showing a more detailed plot containing all of the genes in that profile. |
| − | ##* | + | ##* I took a screenshot of each of the individual profile windows and saved the images in my PowerPoint presentation. |
| − | ##* At the bottom of each profile window, there are two yellow buttons "Profile Gene Table" and "Profile GO Table". For each of the profiles, | + | ##* At the bottom of each profile window, there are two yellow buttons "Profile Gene Table" and "Profile GO Table". For each of the profiles, I clicked on the "Profile Gene Table" button to see the list of genes belonging to the profile. In the window that appeared, I clicked on the "Save Table" button and saved the file to my desktop. I named the file "wt_profile#_genelist.txt", where I replaced the number symbol with the actual profile number. |
| − | ##** | + | ##** I then zipped these files together, and then uploaded these zipped files to the wiki and linked them to this page. |
| − | ##* For each of the significant profiles, | + | ##* For each of the significant profiles, I clicked on the "Profile GO Table" to see the list of Gene Ontology terms belonging to the profile. In the window that appeared, I clicked on the "Save Table" button and saved the file to your desktop. I made the filename "wt_profile#_GOlist.txt" and replaced the number symbol with the actual profile number. |
| − | ##** | + | ##** Again, I zipped these files together, and then uploaded these zipped files to the wiki and linked them to this page. |
| − | |||
# '''Analyzing and Interpreting STEM Results''' | # '''Analyzing and Interpreting STEM Results''' | ||
| − | ## | + | ## I then selected the #45 profile that I had saved in the previous step to further interpret the data. After selecting the profile, I recorded my answers to the following questions. |
| − | ##* '''''Why did you select this profile? | + | ##* '''''Why did you select this profile? In other words, why was it interesting to you?''''' 45 was most interesting to me because it was the most significant profile, meaning it had a p-value of 0.00, and the up-regulation occurs early and then drops. |
##* '''''How many genes belong to this profile?''''' 549.0 | ##* '''''How many genes belong to this profile?''''' 549.0 | ||
##* '''''How many genes were expected to belong to this profile?''''' 47.1 | ##* '''''How many genes were expected to belong to this profile?''''' 47.1 | ||
| − | ##* '''''What is the p value for the enrichment of genes in this profile?''''' | + | ##* '''''What is the p value for the enrichment of genes in this profile?''''' 0.00 |
| − | ## | + | ## I then opened the GO list file I saved for this profile in Excel. This list shows all of the Gene Ontology terms that are associated with genes that fit this profile. I selected the third row and then chose from the menu Data > Filter > Autofilter. I placed a filter on the "p-value" column to show only GO terms that have a p value of < 0.05, and then answered the following question. '''''How many GO terms are associated with this profile at p < 0.05?''''' 81 |
| − | + | ##* The GO list also has a column called "Corrected p-value". This correction is needed because the software has performed thousands of significance tests. I placed a filter on the "Corrected p-value" column to show only GO terms that have a corrected p value of < 0.05, and answered the following question. '''''How many GO terms are associated with this profile with a corrected p value < 0.05?''''' 11 | |
| − | ##* The GO list also has a column called "Corrected p-value". This correction is needed because the software has performed thousands of significance tests. | + | ## I then selected 6 Gene Ontology terms from my filtered list of GO terms with p < 0.05, shown below. |
| − | ## | + | ### nucleolus (GO:0005730) |
| − | ### | + | ### rRNA processing (GO:0006364) |
| − | ### | + | ### ribosome biogenesis (GO:0042254) |
| − | ### | + | ### preribosome, large subunit precursor (GO:0030687) |
| − | ### | + | ### maturation of SSU-rRNA from tricistronic rRNA transcript (SSU-rRNA, 5.8S rRNA, LSU-rRNA)(GO:0000462) |
| − | ### | + | ### RNA binding |
| − | + | ##'''''I then looked up the definitions for each of the terms at [http://geneontology.org http://geneontology.org], and answered the following questions. '''''Why does the cell react to cold shock by changing the expression of genes associated with these GO terms? Also, what does this have to do with the transcription factor being deleted (for the groups working with deletion strain data)?''''' I'm not completely sure why these specific genes had a change in expression, but I noticed that the genes were concerned with regulating RNA and protein production from mRNA. When cold shock is experienced by the cell, there likely needs to be a change in what mRNA is transcribed and at what rate. Additionally, the formation of proteins and the rate at which they are formed must also be regulated. Second question is not related to the wild type strain | |
| − | ### | + | ##* To easily look up the definitions, I went to [http://geneontology.org http://geneontology.org]. |
| − | + | ##* I copy and pasted the GO ID (e.g. GO:0044848) into the search field on the left of the page. | |
| − | ##''''' | + | ##* In the [http://amigo.geneontology.org/amigo/medial_search?q=GO%3A0044848 results] page, I clicked on the button that says "Link to detailed information about <term>, which in this case was "biological phase"". |
| − | ##* To easily look up the definitions, | + | ##* The definition was on the next results page, e.g. [http://amigo.geneontology.org/amigo/term/GO:0044848 here], and the definitions I found are shown below. |
| − | ##* | + | ### definition of nucleolus (GO:0005730): A small, dense body one or more of which are present in the nucleus of eukaryotic cells. It is rich in RNA and protein, is not bounded by a limiting membrane, and is not seen during mitosis. Its prime function is the transcription of the nucleolar DNA into 45S ribosomal-precursor RNA, the processing of this RNA into 5.8S, 18S, and 28S components of ribosomal RNA, and the association of these components with 5S RNA and proteins synthesized outside the nucleolus. This association results in the formation of ribonucleoprotein precursors; these pass into the cytoplasm and mature into the 40S and 60S subunits of the ribosome. |
| − | ##* In the [http://amigo.geneontology.org/amigo/medial_search?q=GO%3A0044848 results] page, | + | ### definition of rRNA processing (GO:0006364): Any process involved in the conversion of a primary ribosomal RNA (rRNA) transcript into one or more mature rRNA molecules. |
| − | ##* The definition | + | ### definition of ribosome biogenesis (GO:0042254): A cellular process that results in the biosynthesis of constituent macromolecules, assembly, and arrangement of constituent parts of ribosome subunits; includes transport to the sites of protein synthesis. |
| − | ### definition of | + | ### definition of preribosome, large subunit precursor (GO:0030687): A preribosomal complex consisting of 27SA, 27SB, and/or 7S pre-rRNA, 5S rRNA, ribosomal proteins including late-associating large subunit proteins, and associated proteins; a precursor of the eukaryotic cytoplasmic large ribosomal subunit. |
| − | ### | + | ### definition of maturation of SSU-rRNA from tricistronic rRNA transcript (SSU-rRNA, 5.8S rRNA, LSU-rRNA)(GO:0000462): Any process involved in the maturation of a precursor Small SubUnit (SSU) ribosomal RNA (rRNA) molecule into a mature SSU-rRNA molecule from the pre-rRNA molecule originally produced as a tricistronic rRNA transcript that contains the Small Subunit (SSU) rRNA, 5.8S rRNA, and the Large Subunit (LSU) in that order from 5' to 3' along the primary transcript. |
| − | + | ### definition of RNA binding (GO:0003723): Binding to an RNA molecule or a portion thereof. | |
| − | ### | ||
| − | ### | ||
| − | ### | ||
| − | |||
| − | |||
| − | |||
==== Using YEASTRACT to Infer which Transcription Factors Regulate a Cluster of Genes (Tuesday, March 26)==== | ==== Using YEASTRACT to Infer which Transcription Factors Regulate a Cluster of Genes (Tuesday, March 26)==== | ||
| − | + | # I then opened the gene list in Excel for the #45 profile. | |
| − | + | #* I copied the list of gene IDs onto my clipboard. | |
| − | + | # I launched a web browser and went to the [http://www.yeastract.com/ YEASTRACT database]. | |
| − | # | + | #* On the left panel of the window, I clicked on the link to [http://www.yeastract.com/formrankbytf.php ''Rank by TF'']. |
| − | #* | + | #* I pasted my list of genes from my cluster into the box labeled ''ORFs/Genes''. |
| − | # | + | #* I checked the box for ''Check for all TFs''. |
| − | #* On the left panel of the window, | + | #* I accepted the defaults for the Regulations Filter (Documented, DNA binding or expression evidence) |
| − | #* | + | #* I did not apply a filter for "Filter Documented Regulations by environmental condition". |
| − | #* | + | #* I ranked the genes by TF using: The % of genes in the list and in YEASTRACT regulated by each TF. |
| − | #* | + | #* I clicked the ''Search'' button. |
| − | #* | + | # Then, I answered the following questions: |
| − | #* | ||
| − | #* | ||
| − | # | ||
#* In the results window that appears, the p values colored green are considered "significant", the ones colored yellow are considered "borderline significant" and the ones colored pink are considered "not significant". '''''How many transcription factors are green or "significant"?''''' 33 | #* In the results window that appears, the p values colored green are considered "significant", the ones colored yellow are considered "borderline significant" and the ones colored pink are considered "not significant". '''''How many transcription factors are green or "significant"?''''' 33 | ||
| − | #* | + | #* I then copied the table of results from the web page and pasted it into a new Excel workbook to preserve the results. |
| − | #** | + | #** I copied by selecting and dragging down on the table. |
| − | #** | + | #** I then uploaded the Excel file to the wiki and linked it in the Data and Files section of this journal assignment. |
| − | + | #** I then answered the following questions. '''''Are CIN5 or GLN3 on the list? If so, what is their "% in user set", "% in YEASTRACT", and "p value"?''''' | |
| − | #** | ||
| − | |||
#*** CIN5 - % in user set: 30.63%, % in scerevisiae: 7.22%, p-value: 0.522044721 | #*** CIN5 - % in user set: 30.63%, % in scerevisiae: 7.22%, p-value: 0.522044721 | ||
#*** GLN3 - % in user set: 38.38%, % in scerevisiae: 8.46%, p-value: 0.002604838 | #*** GLN3 - % in user set: 38.38%, % in scerevisiae: 8.46%, p-value: 0.002604838 | ||
| − | ==== Creating and Visualizing | + | ==== Creating and Visualizing A Gene Regulatory Network with GRNsight (Tuesday, March 26)==== |
| − | + | # I selected from the list of "significant" transcription factors that I found in YEASTRACT to use to run the model. | |
| − | + | #* My chosen transcription factors are shown below. | |
| − | |||
| − | # | ||
| − | #* | ||
#** GCN4 GAT1 RPN4 YAP1 MET28 CBF1 RPH1 MET32 MSN2 STB5 RIM101 MET31 SNF2 SFP1 ASG1 PDR1 CRZ1 MSN4 GLN3 CIN5 | #** GCN4 GAT1 RPN4 YAP1 MET28 CBF1 RPH1 MET32 MSN2 STB5 RIM101 MET31 SNF2 SFP1 ASG1 PDR1 CRZ1 MSN4 GLN3 CIN5 | ||
| − | #** I chose these transcription factors because they were in the top 23 of genes with the smallest p-values, except for GLN3 and CIN5. | + | #** I chose these transcription factors because they were in the top 23 of genes with the smallest p-values, except for GLN3 and CIN5. I added GLN3 and CIN5. |
| − | # | + | # I then went to the [https://dondi.github.io/GRNsight/beta.html GRNsight beta] website. |
| − | # Under the "Network" panel on the left-hand side, | + | # Under the "Network" panel on the left-hand side, I clicked the button "Load from database". |
| − | #* | + | #* I typed the standard name of each transcription factor in the "Select gene" field and clicked the find button (magnifying glass). |
| − | #* | + | #* I continued to add transcription factors in this way until I had added exactly 20 factors, which were the first 18 "significant" factors from my YEASTRACT file and GLN3 and CIN5. |
| − | #* | + | #* I then clicked the "Generate Network" button. |
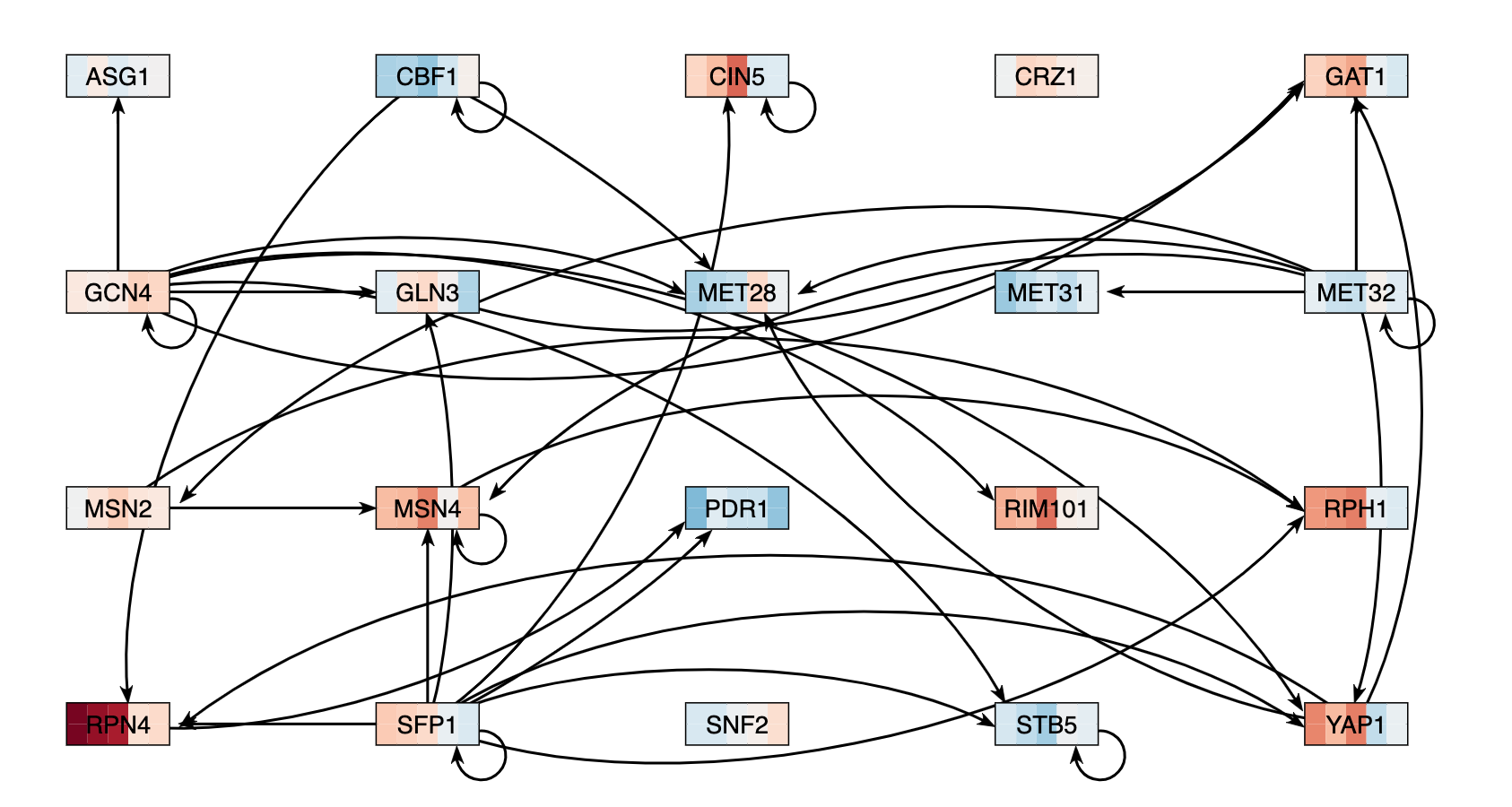
| − | #* | + | #* My network then appeared on the screen, but not every node was connected by at least one arrow to another node. I then repeated the earlier process, but removed the ones that were disconnected and added the next factor from the list. |
| − | + | #* The final network I found had 20 genes and 38 edges | |
| − | #* | + | # Under the "Layout" section, I clicked on the "Grid Layout" button. |
| − | # Under the "Layout" section, | + | #* I exported the network image by going to the Export menu and selecting "Export Image > To PNG", which is shown below and linked in the Data and Files section. |
| − | #* | + | [[image:KM_GRN2.png]] |
==== Creating the GRNmap Input Workbook (Tuesday, March 26)==== | ==== Creating the GRNmap Input Workbook (Tuesday, March 26)==== | ||
| − | + | # With my final network still open in GRNsight, I selected from the Export menu "Export Data > To Excel". In the window that appears, I selected the following: | |
| − | + | #* Under "Select the Expression Data Source:", I chose "Dahlquist_2018" | |
| − | + | #* Under "Select Workbook Sheets to Export:", I selected the following: | |
| − | # With | ||
| − | #* Under "Select the Expression Data Source:", | ||
| − | #* Under "Select Workbook Sheets to Export:", | ||
#** Network sheets | #** Network sheets | ||
#*** "network" | #*** "network" | ||
| Line 135: | Line 114: | ||
#*** "production_rates" | #*** "production_rates" | ||
#*** "threshold_b" | #*** "threshold_b" | ||
| − | #* | + | #* I clicked the "Export Workbook" button. |
| − | # | + | # I opened my workbook in Excel to perform quality control. I checked that it has the following sheets with the following content: |
| − | #* The "network" sheet | + | #* The "network" sheet had an adjacency matrix with my selected regulatory transcription factors across the top row and in the first column. |
| − | #* The "dcin5_log2_expression", "dgln3_log2_expression", and "wt_log2_expression" sheets | + | #* The "dcin5_log2_expression", "dgln3_log2_expression", and "wt_log2_expression" sheets had log2 fold changes for each of your selected regulatory transcription factors for each time point (15, 30, 60, 90, 120). Replicate values had the same column headers. No particular gene was missing all 4-5 replicate values at a particular timepoint for a particular strain, so I did not need to exclude it from the analysis. |
| − | #* The "production_rates" and "degradation_rates" sheets | + | #* The "production_rates" and "degradation_rates" sheets had values for each gene. |
| − | #* The "threshold_b" sheet | + | #* The "threshold_b" sheet had a value of 0 for each gene. |
| − | #* In the "optimization_parameters" sheet, | + | #* In the "optimization_parameters" sheet, I changed the "alpha" value to 0.02 instead of 0.002. |
| − | #* | + | #* I then inserted a new worksheet and named it "network_weights". |
| − | #** | + | #** I copied the entire content of the "network" sheet into the "network_weights" sheet. |
| − | # | + | # I saved and uploaded this Excel Workbook below. |
#* [[Media:KM_GRNmap.xlsx | Excel Workbook KM]] | #* [[Media:KM_GRNmap.xlsx | Excel Workbook KM]] | ||
| − | |||
| − | |||
==Data & Files== | ==Data & Files== | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | [[Media:KM_wt_slide.pptx]] | + | * [[Media:BIOL367_S24_microarray-data_wt_KM2.xlsx | KM WT Workbook]] |
| + | * [[Media:KM_wt_slide.pptx | KM PowerPoint Slide]] | ||
| + | * [[Media:KM_wt_STEM.txt | KM STEM .txt File]] | ||
| + | * [[Media:WT_Profile_GeneList.zip]] [[Media:WT_Profile_GOList.zip]] | ||
| + | * [[Media:KM_GRN2.png | Gene Regulatory Network KM]] | ||
| + | * [[Media:KM_GRN2.xlsx | Excel Workbook for GRN KM]] | ||
==Conclusion== | ==Conclusion== | ||
| + | During this week's journal assignment, I learned how to prepare and download microarray data into STEM. I also gained insight on how to run STEM, and how to view, save, and analyze the results I obtained from the software. I also learned how to use YEASTRACT and how it shows which transcription factors are significant for a given gene expression cluster. I used the list of significant factors I obtained from YEASTRACT to create my Gene Regulatory Network with GRNsight, and then learned how to generate and correct the input workbook for this software. Finally, I worked throughout this process to keep an organized and detailed lab notebook, so that any person could follow these steps and get the same results. | ||
==Acknowledgements== | ==Acknowledgements== | ||
| Line 182: | Line 143: | ||
Except for what is noted above, this individual journal entry was completed by me and not copied from another source. | Except for what is noted above, this individual journal entry was completed by me and not copied from another source. | ||
| − | [[User:Kmill104|Kmill104]] ([[User talk:Kmill104|talk]]) | + | [[User:Kmill104|Kmill104]] ([[User talk:Kmill104|talk]]) 19:33, 10 April 2024 (PDT) |
==References== | ==References== | ||
Latest revision as of 19:33, 10 April 2024
Purpose
The purpose of this week's journal assignment was to analyze a DNA microarray dataset through "high level" analysis. This included using the STEM software for clustering, YEASTRACT for finding which transcription factors regulate these gene clusters, and producing a gene regulatory network in GRNsight. We also worked to keep an organized and detail laboratory notebook, so that another person could follow these steps and reproduce the same results.
Methods/Results
- For this this week's journal assignment, my first step was to prepare my microarray data file for loading into STEM.
- I inserted a new worksheet into my Excel workbook, and named it "wt_stem".
- I then selected all of the data from my "wt_ANOVA" worksheet and Paste special > pasted values into my "wt_stem" worksheet.
- I then renamed the column with the header "Master_Index" to "SPOT". I also renamed the column with the column header "ID" to "Gene Symbol". I deleted the column with the header "Standard_Name".
- I filtered the data on the B-H corrected p value column to be > 0.05.
- Once the data had been filtered, I selected all of the rows (except for my header row) and deleted the rows by right-clicking and choosing "Delete Row" from the context menu. I then undid the filter. This step ensured that the genes I will cluster will only the genes that had a "significant" change in expression.
- I then deleted all of the other data columns EXCEPT for the Average Log Fold change columns for each timepoint.
- I then renamed the data columns with just the time and units, so for any column with wt_AvgLogFC_t#, the header became #m.
- I saved my work. Then, I used Save As to save this spreadsheet as Text (Tab-delimited) (*.txt), clicked OK to the warnings and closed my file.
- Then, I downloaded and extracted the STEM software.
- I clicked on the download link and downloaded the
stem.zipfile to my Desktop. - I extracted the file by right-clicking on it and selecting "Extract all" from the menu.
- This created a folder called
stem.- I then downloaded the Gene Ontology and yeast GO annotations, which I placed in this same folder. I used "gene_ontology.obo" to download the file for Gene Ontology.
- I used "gene_association.sgd.gz" to download the yeast GO annotations.
- Inside the folder, I double-clicked on the
stem.jarto launch the STEM program.
- I clicked on the download link and downloaded the
- Running STEM
- In section 1 (Expression Data Info) of the the main STEM interface window, I clicked on the Browse... button to navigate to and select my file.
- I then clicked on the radio button No normalization/add 0.
- Check the box next to Spot IDs included in the data file.
- In section 2 (Gene Info) of the main STEM interface window, I left the default selection for the three drop-down menu selections for Gene Annotation Source, Cross Reference Source, and Gene Location Source as "User provided".
- I clicked the "Browse..." button to the right of the "Gene Annotation File" item. I then browsed to my "stem" folder and selected the file "gene_association.sgd.gz" and clicked Open.
- In section 3 (Options) of the main STEM interface window, I checked that the Clustering Method says "STEM Clustering Method" and did not change the defaults for Maximum Number of Model Profiles or Maximum Unit Change in Model Profiles between Time Points.
- In section 4 (Execute), I clicked on the yellow Execute button to run STEM.
- In section 1 (Expression Data Info) of the the main STEM interface window, I clicked on the Browse... button to navigate to and select my file.
- Viewing and Saving STEM Results
- A new window opened called "All STEM Profiles (1)". Each box corresponded to a model expression profile. Colored profiles had a statistically significant number of genes assigned; they were arranged in order from most to least significant p value. Profiles with the same color belonged to the same cluster of profiles. The number in each box was simply an ID number for the profile.
- I clicked on the button that says "Interface Options...". At the bottom of the Interface Options window that appears below where it says "X-axis scale should be:", I clicked on the radio button that says "Based on real time". I then closed the Interface Options window.
- I took a screenshot of this window, and then pasted it into a PowerPoint presentation that also contained my p-value table.
- I then clicked on each of the SIGNIFICANT profiles to open a window showing a more detailed plot containing all of the genes in that profile.
- I took a screenshot of each of the individual profile windows and saved the images in my PowerPoint presentation.
- At the bottom of each profile window, there are two yellow buttons "Profile Gene Table" and "Profile GO Table". For each of the profiles, I clicked on the "Profile Gene Table" button to see the list of genes belonging to the profile. In the window that appeared, I clicked on the "Save Table" button and saved the file to my desktop. I named the file "wt_profile#_genelist.txt", where I replaced the number symbol with the actual profile number.
- I then zipped these files together, and then uploaded these zipped files to the wiki and linked them to this page.
- For each of the significant profiles, I clicked on the "Profile GO Table" to see the list of Gene Ontology terms belonging to the profile. In the window that appeared, I clicked on the "Save Table" button and saved the file to your desktop. I made the filename "wt_profile#_GOlist.txt" and replaced the number symbol with the actual profile number.
- Again, I zipped these files together, and then uploaded these zipped files to the wiki and linked them to this page.
- A new window opened called "All STEM Profiles (1)". Each box corresponded to a model expression profile. Colored profiles had a statistically significant number of genes assigned; they were arranged in order from most to least significant p value. Profiles with the same color belonged to the same cluster of profiles. The number in each box was simply an ID number for the profile.
- Analyzing and Interpreting STEM Results
- I then selected the #45 profile that I had saved in the previous step to further interpret the data. After selecting the profile, I recorded my answers to the following questions.
- Why did you select this profile? In other words, why was it interesting to you? 45 was most interesting to me because it was the most significant profile, meaning it had a p-value of 0.00, and the up-regulation occurs early and then drops.
- How many genes belong to this profile? 549.0
- How many genes were expected to belong to this profile? 47.1
- What is the p value for the enrichment of genes in this profile? 0.00
- I then opened the GO list file I saved for this profile in Excel. This list shows all of the Gene Ontology terms that are associated with genes that fit this profile. I selected the third row and then chose from the menu Data > Filter > Autofilter. I placed a filter on the "p-value" column to show only GO terms that have a p value of < 0.05, and then answered the following question. How many GO terms are associated with this profile at p < 0.05? 81
- The GO list also has a column called "Corrected p-value". This correction is needed because the software has performed thousands of significance tests. I placed a filter on the "Corrected p-value" column to show only GO terms that have a corrected p value of < 0.05, and answered the following question. How many GO terms are associated with this profile with a corrected p value < 0.05? 11
- I then selected 6 Gene Ontology terms from my filtered list of GO terms with p < 0.05, shown below.
- nucleolus (GO:0005730)
- rRNA processing (GO:0006364)
- ribosome biogenesis (GO:0042254)
- preribosome, large subunit precursor (GO:0030687)
- maturation of SSU-rRNA from tricistronic rRNA transcript (SSU-rRNA, 5.8S rRNA, LSU-rRNA)(GO:0000462)
- RNA binding
- I then looked up the definitions for each of the terms at http://geneontology.org, and answered the following questions. Why does the cell react to cold shock by changing the expression of genes associated with these GO terms? Also, what does this have to do with the transcription factor being deleted (for the groups working with deletion strain data)? I'm not completely sure why these specific genes had a change in expression, but I noticed that the genes were concerned with regulating RNA and protein production from mRNA. When cold shock is experienced by the cell, there likely needs to be a change in what mRNA is transcribed and at what rate. Additionally, the formation of proteins and the rate at which they are formed must also be regulated. Second question is not related to the wild type strain
- To easily look up the definitions, I went to http://geneontology.org.
- I copy and pasted the GO ID (e.g. GO:0044848) into the search field on the left of the page.
- In the results page, I clicked on the button that says "Link to detailed information about <term>, which in this case was "biological phase"".
- The definition was on the next results page, e.g. here, and the definitions I found are shown below.
- definition of nucleolus (GO:0005730): A small, dense body one or more of which are present in the nucleus of eukaryotic cells. It is rich in RNA and protein, is not bounded by a limiting membrane, and is not seen during mitosis. Its prime function is the transcription of the nucleolar DNA into 45S ribosomal-precursor RNA, the processing of this RNA into 5.8S, 18S, and 28S components of ribosomal RNA, and the association of these components with 5S RNA and proteins synthesized outside the nucleolus. This association results in the formation of ribonucleoprotein precursors; these pass into the cytoplasm and mature into the 40S and 60S subunits of the ribosome.
- definition of rRNA processing (GO:0006364): Any process involved in the conversion of a primary ribosomal RNA (rRNA) transcript into one or more mature rRNA molecules.
- definition of ribosome biogenesis (GO:0042254): A cellular process that results in the biosynthesis of constituent macromolecules, assembly, and arrangement of constituent parts of ribosome subunits; includes transport to the sites of protein synthesis.
- definition of preribosome, large subunit precursor (GO:0030687): A preribosomal complex consisting of 27SA, 27SB, and/or 7S pre-rRNA, 5S rRNA, ribosomal proteins including late-associating large subunit proteins, and associated proteins; a precursor of the eukaryotic cytoplasmic large ribosomal subunit.
- definition of maturation of SSU-rRNA from tricistronic rRNA transcript (SSU-rRNA, 5.8S rRNA, LSU-rRNA)(GO:0000462): Any process involved in the maturation of a precursor Small SubUnit (SSU) ribosomal RNA (rRNA) molecule into a mature SSU-rRNA molecule from the pre-rRNA molecule originally produced as a tricistronic rRNA transcript that contains the Small Subunit (SSU) rRNA, 5.8S rRNA, and the Large Subunit (LSU) in that order from 5' to 3' along the primary transcript.
- definition of RNA binding (GO:0003723): Binding to an RNA molecule or a portion thereof.
- I then selected the #45 profile that I had saved in the previous step to further interpret the data. After selecting the profile, I recorded my answers to the following questions.
Using YEASTRACT to Infer which Transcription Factors Regulate a Cluster of Genes (Tuesday, March 26)
- I then opened the gene list in Excel for the #45 profile.
- I copied the list of gene IDs onto my clipboard.
- I launched a web browser and went to the YEASTRACT database.
- On the left panel of the window, I clicked on the link to Rank by TF.
- I pasted my list of genes from my cluster into the box labeled ORFs/Genes.
- I checked the box for Check for all TFs.
- I accepted the defaults for the Regulations Filter (Documented, DNA binding or expression evidence)
- I did not apply a filter for "Filter Documented Regulations by environmental condition".
- I ranked the genes by TF using: The % of genes in the list and in YEASTRACT regulated by each TF.
- I clicked the Search button.
- Then, I answered the following questions:
- In the results window that appears, the p values colored green are considered "significant", the ones colored yellow are considered "borderline significant" and the ones colored pink are considered "not significant". How many transcription factors are green or "significant"? 33
- I then copied the table of results from the web page and pasted it into a new Excel workbook to preserve the results.
- I copied by selecting and dragging down on the table.
- I then uploaded the Excel file to the wiki and linked it in the Data and Files section of this journal assignment.
- I then answered the following questions. Are CIN5 or GLN3 on the list? If so, what is their "% in user set", "% in YEASTRACT", and "p value"?
- CIN5 - % in user set: 30.63%, % in scerevisiae: 7.22%, p-value: 0.522044721
- GLN3 - % in user set: 38.38%, % in scerevisiae: 8.46%, p-value: 0.002604838
Creating and Visualizing A Gene Regulatory Network with GRNsight (Tuesday, March 26)
- I selected from the list of "significant" transcription factors that I found in YEASTRACT to use to run the model.
- My chosen transcription factors are shown below.
- GCN4 GAT1 RPN4 YAP1 MET28 CBF1 RPH1 MET32 MSN2 STB5 RIM101 MET31 SNF2 SFP1 ASG1 PDR1 CRZ1 MSN4 GLN3 CIN5
- I chose these transcription factors because they were in the top 23 of genes with the smallest p-values, except for GLN3 and CIN5. I added GLN3 and CIN5.
- My chosen transcription factors are shown below.
- I then went to the GRNsight beta website.
- Under the "Network" panel on the left-hand side, I clicked the button "Load from database".
- I typed the standard name of each transcription factor in the "Select gene" field and clicked the find button (magnifying glass).
- I continued to add transcription factors in this way until I had added exactly 20 factors, which were the first 18 "significant" factors from my YEASTRACT file and GLN3 and CIN5.
- I then clicked the "Generate Network" button.
- My network then appeared on the screen, but not every node was connected by at least one arrow to another node. I then repeated the earlier process, but removed the ones that were disconnected and added the next factor from the list.
- The final network I found had 20 genes and 38 edges
- Under the "Layout" section, I clicked on the "Grid Layout" button.
- I exported the network image by going to the Export menu and selecting "Export Image > To PNG", which is shown below and linked in the Data and Files section.

Creating the GRNmap Input Workbook (Tuesday, March 26)
- With my final network still open in GRNsight, I selected from the Export menu "Export Data > To Excel". In the window that appears, I selected the following:
- Under "Select the Expression Data Source:", I chose "Dahlquist_2018"
- Under "Select Workbook Sheets to Export:", I selected the following:
- Network sheets
- "network"
- Expression sheets
- dcin5_log2_expression
- dgln3_log2_expression
- wt_log2_expression
- Additional sheets
- "degradation_rates"
- "optimization_parameters"
- "production_rates"
- "threshold_b"
- Network sheets
- I clicked the "Export Workbook" button.
- I opened my workbook in Excel to perform quality control. I checked that it has the following sheets with the following content:
- The "network" sheet had an adjacency matrix with my selected regulatory transcription factors across the top row and in the first column.
- The "dcin5_log2_expression", "dgln3_log2_expression", and "wt_log2_expression" sheets had log2 fold changes for each of your selected regulatory transcription factors for each time point (15, 30, 60, 90, 120). Replicate values had the same column headers. No particular gene was missing all 4-5 replicate values at a particular timepoint for a particular strain, so I did not need to exclude it from the analysis.
- The "production_rates" and "degradation_rates" sheets had values for each gene.
- The "threshold_b" sheet had a value of 0 for each gene.
- In the "optimization_parameters" sheet, I changed the "alpha" value to 0.02 instead of 0.002.
- I then inserted a new worksheet and named it "network_weights".
- I copied the entire content of the "network" sheet into the "network_weights" sheet.
- I saved and uploaded this Excel Workbook below.
Data & Files
- KM WT Workbook
- KM PowerPoint Slide
- KM STEM .txt File
- Media:WT_Profile_GeneList.zip Media:WT_Profile_GOList.zip
- Gene Regulatory Network KM
- Excel Workbook for GRN KM
{kind=link}
Conclusion
During this week's journal assignment, I learned how to prepare and download microarray data into STEM. I also gained insight on how to run STEM, and how to view, save, and analyze the results I obtained from the software. I also learned how to use YEASTRACT and how it shows which transcription factors are significant for a given gene expression cluster. I used the list of significant factors I obtained from YEASTRACT to create my Gene Regulatory Network with GRNsight, and then learned how to generate and correct the input workbook for this software. Finally, I worked throughout this process to keep an organized and detailed lab notebook, so that any person could follow these steps and get the same results.
Acknowledgements
I worked under the guidance of Dr. Dahlquist on 3-21-24 and 3-26-24. I either consulted Dr. Dahlquist or my classmates if I ever had questions about using STEM, YEASTRACT, or GRNsight. For the procedure section of this individual journal, I first copied the general procedure from the Week 10 assignment page, and then adjusted it so that the specific steps I followed are described above.
Except for what is noted above, this individual journal entry was completed by me and not copied from another source.
Kmill104 (talk) 19:33, 10 April 2024 (PDT)
References
- Dahlquist, K. D., Fitzpatrick, B. G., Camacho, E. T., Entzminger, S. D., & Wanner, N. C. (2015). Parameter estimation for gene regulatory networks from microarray data: cold shock response in Saccharomyces cerevisiae. Bulletin of mathematical biology, 77(8), 1457-1492. DOI: 10.1007/s11538-015-0092-6
- Ernst, J., & Bar-Joseph, Z. (2006). STEM: a tool for the analysis of short time series gene expression data. BMC bioinformatics, 7(1), 191. DOI: 10.1093/bioinformatics/bti1022
- LMU BioDB 2024. (2024). Week 10. Retrieved April 2, 2024, from https://xmlpipedb.cs.lmu.edu/biodb/spring2024/index.php/Week_10
- Teixeira, M. C., Viana, R., Palma, M., Oliveira, J., Galocha, M., Mota, M. N., ... & Monteiro, P. T. (2023). YEASTRACT+: a portal for the exploitation of global transcription regulation and metabolic model data in yeast biotechnology and pathogenesis. Nucleic Acids Research, 51(D1), D785-D791. DOI: 10.1093/nar/gkac1041
User Page
Assignment Pages
- Week 1
- Week 2
- Week 3
- Week 4
- Week 5
- Week 6
- Week 8
- Week 9
- Week 10
- Week 11
- Week 12
- Week 13
- Week 14
- Week 15
Individual Journal Entry Pages
- Kmill104 Week 1
- Kmill104 Week 2
- MSymond1 KMill104 Week 3
- Monarch Initiative Week 4
- Kmill104 Week 5
- Kmill104 Week 6
- Kmill104 Week 8
- Kmill104 Week 9
- Kmill104 Week 10
- Kmill104 Week 11
- Kmill104 Week 12
- Data Analysts Week 13
- Data Analysts Week 14
- Data Analysts Week 15