Bklein7 Week 8
Contents
- 1 Files Generated in the This Week's Analysis
- 2 Statistical Analysis of Vibrio cholerae Microarray Data (Part 1)
- 3 MAPPFinder Analysis of Vibrio cholerae Microarray Data (Part 2)
- 4 Conclusion
- 5 Links
Files Generated in the This Week's Analysis
It may be easier to zip all of these files together and then upload them as a single zipped file, rather than zipping and uploading individually (for filetypes not allowed by OpenWetware).
- Your exceptions file when you imported your data into GenMAPP:
.EX.txt - Your Expression Dataset file:
.gex
- Current .gex File: File:Merrell Compiled Raw Data Vibrio BK 20151015.gex
- Your GO results file:
XXX-CriterionX-GO.txt
- Your GO results saved as an Excel spreadsheet with filters applied:
.xls
- The MAPP you looked at:
.mapp
- The MAPPFinder GO mappings file:
.gmf
- Part I Spreadsheet: File:Merrell Compiled Raw Data Vibrio BK 20151015.xls
- Part I tab delimited File: File:Merrell Compiled Raw Data Vibrio BK 20151015- Tab Delimited.txt
- Current .gex File: File:Merrell Compiled Raw Data Vibrio BK 20151015.gex
- File:Merrell Compiled Raw Data Vibrio BK 20151015-Criterion1-GO.txt

- File:3’-5’-exoribonuclease activity.mapp


Statistical Analysis of Vibrio cholerae Microarray Data (Part 1)
- Attaining the unanalyzed Vibrio cholerae DNA microarray data
- The Vibrio cholerae data was accessed by downloading the following file: File:Merrell Compiled Raw Data Vibrio.xls.
- The above file was renamed to File:Merrell Compiled Raw Data Vibrio BK 20151015.xls for the purposes of this section.
- The analysis performed on the Vibrio cholera is detailed in the sections below:
Normalizing the Log Ratios for the Set of Slides in the Experiment
This section dictates the steps necessary to scale and center the raw microarray data:
- To begin, I created a new Worksheet in my Excel file entitled "scaled_centered".
- I went back to the original "compiled_raw_data" worksheet and copied over all the data into the new "scaled_centered" worksheet.
- I inserted two rows in between the top row of headers and the first data row entitled "Average" (cell A2) "StdDev" (cell A3)
- I computed the Average log ratio for each chip by inputting the following equation into cell B2 and then pasting it into the rest of the "Average" column:
=AVERAGE(B4:B5224)
- I computed the Standard Deviation of the log ratios on each chip by inputting the following equation into cell B3 and then pasting it into the rest of the "StdDev" column:
=STDEV(B4:B5224)
- I created a new set of headings for the scaled and centered data by copying over the data column headings and then pasting them to the right of the last data column. I edited the names of the columns so that they now read: A1_scaled_centered, A2_scaled_centered, etc.
- In cell N4 (column with the heading A1_scaled_centered), I typed the following equation:
=(B4-B$2)/B$3
- In this case, we want the data in cell B4 to have the average subtracted from it (cell B2) and be divided by the standard deviation (cell B3). We use the dollar sign symbols in front of the "2" and "3" to tell Excel to always reference that row in the equation, even though we will paste it for the entire column of 5221 genes. Why is this important?
- I copied this equation to the rest of the column and then adapted it for all "_scaled_centered" columns.
Performing Statistical Analysis on the Ratios
This section details the steps necessary to perform statistical analysis on the scaled and centered data produced in the section above:
- For this section, I created a new worksheet and name it "statistics".
- I went back to the "scaled_centered" worksheet and copied over both the first column ("ID") and copied the columns that are designated "_scaled_centered".
- I deleted rows 2 and 3 where it says "Average" and "StDev" so that the data rows with gene IDs were immediately below the header row 1.
- I created three new columns to the right of the copied ones with the following headers: "Avg_LogFC_A", "Avg_LogFC_B", and "Avg_LogFC_C".
- I computed the average log fold change for the replicates for each patient by typing the equation:
=AVERAGE(B2:E2)
- into cell N2 and then copying/pasting it into the rest of the three columns, adapting it as necessary.
- I created a new column to compute the average of the averages with the header "Avg_LogFC_all". I then created the equation to compute the average of the three previous averages you calculated and paste it into this entire column.
- I created a new column with the header "Tstat" to compute the T statistic for each scaled and centered average log ratio. In this column, I entered the following equation and pasted it into the rest of the column:
=AVERAGE(N2:P2)/(STDEV(N2:P2)/SQRT(3))
- I created one last column with the heading "Pvalue". In the cell below the label, I entered the following equation and then copied it into the rest of the column:
=TDIST(ABS(R2),2,2)
Calculating the Bonferroni p value Correction
It is necessary to perform adjustments to the p value to correct for the multiple testing problem. Therefore, I first calculated the Bonferroni p value correction on the p values calculated in the above section:
- I labelled the next two columns to the right of "Pvalue" with the same label: "Bonferroni_Pvalue".
- I typed the equation
=S2*5221into the first Bonferroni column and pasted it throughout. - Next, I replaced any corrected p value that was greater than 1 with the number 1 by typing the following formula into the first cell below the second Bonferroni_Pvalue header:
=IF(T2>1,1,T2).
Calculating the Benjamini & Hochberg p value Correction
The second p value correction I performed was the Benjamini & Hochberg correction, the methods of which are presented below:
- I created a new worksheet named "B-H_Pvalue".
- In this worksheet, I copied over the "ID" column from the previous worksheet into the first column of the new worksheet.
- Next, I added a new column on the very left and named it "MasterIndex". Under this heading, I added a numbered list from 1 to 5221 (the number of genes on the microarray).
- I copied over the unadjusted p values from your previous worksheet and pasted them into Column C.
- I selected all of columns A, B, and C and sorted by ascending values on Column C.
- This was done by clicking the sort button from A to Z on the toolbar and sorting by column C, smallest to largest.
- I created a new column and labelled it with the header "Rank" in cell D1 to house another numbered list from 1 to 5221. Because this was done for the sorted columns, these values represented the p value ranks, smallest to largest.
- I created a two new columns to calculate the Benjamini and Hochberg p value correction and labeled them "B-H_Pvalue" (cell E1 and F1).
- In the first column, I inputted the following formula and copied it throughout the column:
=(C2*5221)/D2. - In the second column, I inputted the following formula and copied it throughout:
=IF(E2>1,1,E2).
- In the first column, I inputted the following formula and copied it throughout the column:
- Next, I selected columns A through F and sorted them by the MasterIndex in Column A in ascending order.
- Finally, I copied column F into the next column on the right of your "statistics" sheet.
Preparing the File for GenMAPP
- I inserted a new worksheet and named it "forGenMAPP".
- I copied over the entirety of the "statistics" worksheet to this new worksheet.
- I selected Columns B through Q (all the fold changes) and formatted the cells to show only 2 decimal places.
- I selected all the columns containing p values and formatted the cells to show only 4 decimal places.
- I deleted the left-most Bonferroni p value column.
- I inserted a column to the right of the "ID" column entitled "SystemCode" and filled the entire column with the letter "N".
- After making the above changes, I saved the file as "Text (Tab-delimited) (*.txt)".
- This .txt file can be found here: File:Merrell Compiled Raw Data Vibrio BK 20151015- Tab Delimited.txt.
Sanity Check: Number of Genes Significantly Changed
To verify the results of the data analysis performed on the Vibrio cholerae DNA microarray data, I assessed the number of genes that were significantly changed at various p value cut-offs and also compared my data analysis with the published results of Merrell et al. (2002). These analyses were done in the "forGenMapp" tab of the Excel spreadsheet used for the data analysis.
- Assessing the number of genes significantly changed
- To answer the questions below, I used custom filter to display only the "Pvalue" data that met specific criterion (e.g. < 0.05).
- How many genes have p value < 0.05? and what is the percentage (out of 5221)?
- What about p < 0.01? and what is the percentage (out of 5221)?
- What about p < 0.001? and what is the percentage (out of 5221)?
- What about p < 0.0001? and what is the percentage (out of 5221)?
- ((We have just performed 5221 T tests for significance. Another way to state what we are seeing with p < 0.05 is that we would expect to see this magnitude of a gene expression change in about 5% of our T tests, or 261 times. Since we have more than 261 genes that pass this cut off, we know that some genes are significantly changed. However, we don't know which ones. To apply a more stringent criterion to our p values, we performed the Bonferroni and Benjamini and Hochberg corrections to these unadjusted p values. The Bonferroni correction is very stringent. The Benjamini-Hochberg correction is less stringent. To see this relationship, filter your data to determine the following:))
- How many genes are p < 0.05 for the Bonferroni-corrected p value? and what is the percentage (out of 5221)?
- How many genes are p < 0.05 for the Benjamini and Hochberg-corrected p value? and what is the percentage (out of 5221)?
- Assessing the magnitude and direction of significant gene expression changes
- The "Avg_LogFC_all" indicated the size of the gene expression changes and their direction. Positive values increased relative to the control, and negative values decreased relative to the control.
- Keeping the (unadjusted) "Pvalue" filter at p < 0.05, I filtered the "Avg_LogFC_all" column to show all genes with an average log fold change greater than zero. How many are there? (and %)
- Keeping the (unadjusted) "Pvalue" filter at p < 0.05, I filtered the "Avg_LogFC_all" column to show all genes with an average log fold change less than zero. How many are there? (and %)
- What about an average log fold change of > 0.25 and p < 0.05? (and %)
- Or an average log fold change of < -0.25 and p < 0.05? (and %)
- The "Avg_LogFC_all" indicated the size of the gene expression changes and their direction. Positive values increased relative to the control, and negative values decreased relative to the control.
- What criteria did Merrell et al. (2002) use to determine a significant gene expression change? How does it compare to our method?
- For the GenMAPP analysis below, I used the fold change cut-off of greater than 0.25 or less than -0.25 and the unadjusted p value cut off of p < 0.05 for the analysis.
Sanity Check: Compare Individual Genes with Known Data
Merrell et al. (2002) report that genes with IDs: VC0028, VC0941, VC0869, VC0051, VC0647, VC0468, VC2350, and VCA0583 were all significantly changed in their data. I reviewed the "forGenMAPP" Excel worksheet to compare these findings to the results of my personal data analysis.
- What are the fold changes and p values (of these genes)? Are they significantly changed in the analysis?
- VC0028
- VC0941
- VC0869
- VC0051
- VC0647
- VC0468
- VC2350
- VCA0583
MAPPFinder Analysis of Vibrio cholerae Microarray Data (Part 2)
Mapping Onto Biological Pathways (GenMAPP & MAPPFinder)
Before beginning the mapping process, it is necessary to load the correct gene database into MAPPFinder:
- I download the 2009 Vibrio cholerae gene database by following this link to the XMLPipeDB SourceForge Download page. My homework partner Veronica downloaded the 2010 version.
- I saved the above file into the folder C:\GenMAPP 2 Data\Gene Databases and extracted it.
- Within MAPPFinder, I loaded the 2009 gene database by selecting Data > Choose Gene Database and choosing the appropriate file from the directory C:\GenMAPP 2 Data\Gene Databases.
GenMAPP Expression Dataset Manager Procedure
- Launch the GenMAPP Program. Check to make sure the correct Gene Database is loaded.
- Look in the lower, left-hand corner of the main GenMAPP Drafting Board window to see the name of the Gene Database that is loaded. If this is not the correct Gene Database or it says "No Gene Database", then go to the Data > Choose Gene Database menu item to select the Gene Database you need to perform the analysis.
- Remember, you and your partner are going to use different versions of the Vibrio cholerae Gene Database for this exercise.
- Select the Data menu from the main Drafting Board window and choose Expression Dataset Manager from the drop-down list. The Expression Dataset Manager window will open.
- Select New Dataset from the Expression Datasets menu. Select the tab-delimited text file that you formatted for GenMAPP (.txt) in the procedure above from the file dialog box that appears.
- You may need to download your .txt file from the wiki onto your Desktop if you have not already done so.
- The Data Type Specification window will appear. GenMAPP is expecting that you are providing numerical data. If any of your columns has text (character) data, you would check the box next to the field (column) name.
- The Vibrio data we have been working with does not have any text (character) data in it.
- Allow the Expression Dataset Manager to convert your data.
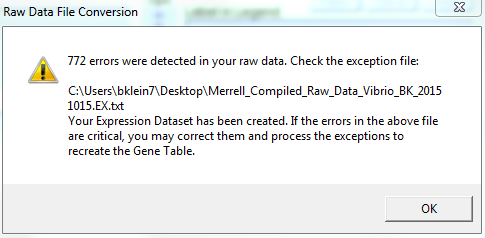
- This may take a few minutes depending on the size of the dataset and the computer’s memory and processor speed. When the process is complete, the converted dataset will be active in the Expression Dataset Manager window and the file will be saved in the same folder the raw data file was in, named the same except with a .gex extension; for example, MyExperiment.gex.
- NOTE: "772 ERORRS WERE DETECTED IN YOUR RAW DATA... YOUR EXPRESSION DATASET HAS BEEN CREATED. IF THE ERRORS IN THE ABOVE FILE ARE CRITICAL, YOU MAY CORRECT THEM AND PROCESS THE EXCEPTIONS TO RECREATE THE GENE TABLE" VS. 121 ERRORS FOR VERONICA
- A message may appear saying that the Expression Dataset Manager could not convert one or more lines of data. Lines that generate an error during the conversion of a raw data file are not added to the Expression Dataset. Instead, an exception file is created. The exception file is given the same name as your raw data file with .EX before the extension (e.g., MyExperiment.EX.txt). The exception file will contain all of your raw data, with the addition of a column named ~Error~. This column contains either error messages or, if the program finds no errors, a single space character.
- Record the number of errors. For your journal assignment, open the .EX.txt file and use the Data > Filter > Autofilter function to determine what the errors were for the rows that were not converted. Record this information in your individual journal page.
- It is likely that you will have a different number of errors than your partner who is using a different version of the Vibrio cholerae Gene Database. Which of you has more errors? Why do you think that is? Record your answers in your journal page.
- Upload your exceptions file:
EX.txtto your wiki page.
- Customize the new Expression Dataset by creating new Color Sets which contain the instructions to GenMAPP for displaying data on MAPPs.
- Color Sets contain the instructions to GenMAPP for displaying data from an Expression Dataset on MAPPs. Create a Color Set by filling in the following different fields in the Color Set area of the Expression Dataset Manager: a name for the Color Set, the gene value, and the criteria that determine how a gene object is colored on the MAPP. Enter a name in the Color Set Name field that is 20 characters or fewer.
- The Gene Value is the data displayed next to the gene box on a MAPP. Select the column of data to be used as the Gene Value from the drop down list or select [none]. We will use "Avg_LogFC_all" for the Vibrio dataset you just created.
- Activate the Criteria Builder by clicking the New button.
- Enter a name for the criterion in the Label in Legend field.
- Choose a color for the criterion by left-clicking on the Color box. Choose a color from the Color window that appears and click OK.
- State the criterion for color-coding a gene in the Criterion field.
- A criterion is stated with relationships such as "this column greater than this value" or "that column less than or equal to that value". Individual relationships can be combined using as many ANDs and ORs as needed. A typical relationship is
[ColumnName] RelationalOperator Value
- with the column name always enclosed in brackets and character values enclosed in single quotes. For example:
[Fold Change] >= 2 [p value] < 0.05 [Quality] = 'high'
- This is the equivalent to queries that you performed on the command line when working with the PostgreSQL movie database. GenMAPP is using a graphical user interface (GUI) to help the user format the queries correctly. The easiest and safest way to create criteria is by choosing items from the Columns and Ops (operators) lists shown in the Criteria Builder. The Columns list contains all of the column headings from your Expression Dataset. To choose a column from the list, click on the column heading. It will appear at the location of the cursor in the Criterion box. The Criteria Builder surrounds the column names with brackets.
- The Ops (operators) list contains the relational operators that may be used in the criteria: equals ( = ) greater than ( > ), less than ( < ), greater than or equal to ( >= ), less than or equal to ( <= ), is not equal to ( <> ). To choose an operator from the list, click on the symbol. It will appear at the location of the insertion bar (cursor) in the Criterion box. The Criteria Builder automatically surrounds the operators with spaces.
- The Ops list also contains the conjunctions AND and OR, which may be used to make compound criteria. For example:
[Fold Change] > 1.2 AND [p value] <= 0.05
- Parentheses control the order of evaluation. Anything in parentheses is evaluated first. Parentheses may be nested. For example:
[Control Average] = 100 AND ([Exp1 Average] > 100 OR [Exp2 Average] > 100)
- Column names may be used anywhere a value can, for example:
[Control Average] < [Experiment Average]
- After completing a new criterion, add the criterion entry (label, criterion, and color) to the Criteria List by clicking the Add button.
- For the Vibrio dataset, you will create two criterion. "Increased" will be [Avg_LogFC_all] > 0.25 AND [Pvalue] < 0.05 and "Decreased will be [Avg_LogFC_all] < -0.25 AND [Pvalue] < 0.05.
- You may continue to add criteria to the Color Set by using the previous steps.
- The buttons to the right of the list represent actions that can be performed on individual criteria. To modify a criterion label, color, or the criterion itself, first select the criterion in the list by left-clicking on it, and then click the Edit button. This puts the selected criterion into the Criteria Builder to be modified. Click the Save button to save changes to the modified criterion; click the Add button to add it to the list as a separate criterion. To remove a criterion from the list, left-click on the criterion to select it, and then click on the Delete button. The order of Criteria in the list has significance to GenMAPP. When applying an Expression Dataset and Color Set to a MAPP, GenMAPP examines the expression data for a particular gene object and applies the color for the first criterion in the list that is true. Therefore, it is imperative that when criteria overlap the user put the most important or least inclusive criteria in the list first. To change the order of the criteria in the list, left-click on the criterion to select it and then click the Move Up or Move Down buttons. No criteria met and Not found are always the last two positions in the list.
- Save the entire Expression Dataset by selecting Save from the Expression Dataset menu. Changes made to a Color Set are not saved until you do this.
- Exit the Expression Dataset Manager to view the Color Sets on a MAPP. Choose Exit from the Expression Dataset menu or click the close box in the upper right hand corner of the window.
- Upload your .gex file to your journal entry page for later retrieval.
MAPPFinder Procedure
Note: You and your partner will both do the same criterion, either "Increased" or "Decreased", but your group does not need to do both "Increased" and "Decreased" Sign up for the criterion you want on the group list ( Fall 2010 or Fall 2013) so that we can make sure that as a class we are covering both criteria.
- Launch the MAPPFinder program (or from within GenMAPP, select Tools > MAPPFinder).
- Make sure that the Gene Database for the correct species is loaded. The name of the Gene Database appears at the bottom of the window. If this is not the right one, go to File > Choose Gene Database and choose the correct one. (The Gene Databases are stored in the folder C:\GenMAPP 2 Data\Gene Databases\.)
- Click on the button "Calculate New Results".
- Click on "Find File" and choose the your Expression Dataset file, for example, "MyDataset.gex", and click OK.
- MAPPFinder may have found it for you already if you already had it open in GenMAPP, in which case, you just need to click OK.
- Choose the Color Set and Criteria with which to filter the data. Click on either the "Increased" and "Decreased" criteria in the right-hand box, depending on which one your group is doing. (You could select both by holding down the Control key while clicking).
- Check the boxes next to "Gene Ontology" and "p value".
- Click the "Browse" button and create a meaningful filename for your results.
- Click "Run MAPPFinder". The analysis will take several minutes. It may look like the computer is stalled; be patient, it will eventually start running.
- When the results have been calculated, a Gene Ontology browser will open showing your results. All of the Gene Ontology terms that have at least 3 genes measured and a p value of less than 0.05 will be highlighted yellow. A term with a p value less than 0.05 is considered a "significant" result. Browse through the tree to see your results.
- To see a list of the most significant Gene Ontology terms, click on the menu item "Show Ranked List".
- List the top 10 Gene Ontology terms in your individual journal entry.
- Compare your list with your partner who used a different version of the Gene Database. Are your terms the same or different? Why do you think that is? Record your answer in your individual journal entry.
- One of the things you can do in MAPPFinder is to find the Gene Ontology term(s) with which a particular gene is associated. First, in the main MAPPFinder Browser window, click on the button "Collapse the Tree". Then, you can search for the genes that were mentioned by Merrell et al. (2002), VC0028, VC0941, VC0869, VC0051, VC0647, VC0468, VC2350, and VCA0583. Type the identifier for one of these genes into the MAPPFinder browser gene ID search field. Choose "OrderedLocusNames" from the drop-down menu to the right of the search field. Click on the GeneID Search button. The GO term(s) that are associated with that gene will be highlighted in blue. List the GO terms associated with each of those genes in your individual journal. (Note: they might not all be found.) Are they the same as your partner who is using a different Gene Database? Why or why not?
- Click on one of the GO terms that are associated with one of the genes you looked up in the previous step. A MAPP will open listing all of the genes (as boxes) associated with that GO term. The genes named within the map are based on the UniProt identification system. To match the gene of interest to its identification go to the UniProt site and type in your gene ID into the search bar. Moreover, the genes on the MAPP will be color-coded with the gene expression data from the microarray experiment. List in your journal entry the name of the GO term you clicked on and whether the expression of the gene you were looking for changed significantly in the experiment.
- Double-click on the gene box. This will open a Internet Explorer window called the "Backpage" for this gene. This page has links to pages for this gene in the public databases. Click on the links to find out the function of this gene and record your answer in your individual journal page.
- The MAPP that has just been created is stored in the directory, C:\GenMAPP 2 Data\MAPPs\VC GO. Upload this file and link to it in your journal.
- In Windows, make a copy of your results (XXX-CriterionX-GO.txt) file.
- "XXX" refers to the name you gave to your results file.
- "CriterionX" refers to either "Criterion0" or "Criterion1". Since computers start counting at zero, "Criterion0" is the first criterion in the list you clicked on ("Increased" if you followed the directions) and "Criterion1" is the second criterion in the list you clicked on ("Decreased" if you followed the directions).
- Upload your results file to your journal page.
- Launch Microsoft Excel. Open the copies of the .txt files in Excel (you will need to "Show all files" and click "Finish" to the wizard that will open your file). This will show you the same data that you saw in the MAPPFinder Browser, but in tabular form.
- Look at the top of the spreadsheet. There are rows of information that give you the background information on how MAPPFinder made the calculations. Compare this information with your partner who used a different version of the Vibrio Gene Database. Which numbers are different? Why are they different? Record this information in your individual journal entry.
- You will filter this list to show the top GO terms represented in your data for both the "Increased" and "Decreased" criteria. You will need to filter your list down to about 20 terms. Click on a cell in the row of headers for the data. Then go to the Data menu and click "Filter > Autofilter". Drop-down arrows will appear in the row of headers. You can now choose to filter the data. Click on the drop-down arrow for the column you wish to filter and choose "(Custom…)". A window will open giving you choices on how you want to filter. You must set these two filters:
Z Score (in column N) greater than 2 PermuteP (in column O) less than 0.05
- You will use these two filters depending on the number of terms you have:
Number Changed (in column I) greater than or equal to 4 or 5 AND less than 100 Percent Changed (in column L) greater than or equal to 25-50%
- Save your changes to an Excel spreadsheet. Select File > Save As and select Excel workbook (.xls) from the drop-down menu. Your filter settings won’t be saved in a .txt file.
- Are any of your filtered GO terms closely related to one another, meaning are they a direct child or parent to another term in the list? You can judge this by comparing your spreadsheet with the MAPPFinder browser. Highlight the terms that fit this relationship with the same color in your Excel spreadsheet. Upload your .xls file to your journal page.
- Interpret your results. Look up the definitions for any GO terms that are unfamiliar to you. The "official" definitions for GO terms can be found at http://www.geneontology.org. You can use one of the online biological dictionaries as a supplement, if needed. Write a paragraph relating the results of this GO analysis to the experiment performed (comparing laboratory-grown and patient-derived Vibrio cholerae. You need to give a biological interpretation of what do each of these GO terms in your filtered list have to to with the pathogenecity of the bacterium? You may consult with your partner on this, but your explanation on your individual journal page needs to be in your own words. This is where the real "brain power" comes in with interpreting DNA microarray data. Even experienced scientists struggle with this part. Use your creativity as a scientist to stretch your brain in this question.
- There is one other file you need to save to your journal page. It has a .gmf extension and should be in the same fold as the .gex file that you created with the GenMAPP Expression Dataset Manager. You will need this file to re-open your results in MAPPFinder.
Conclusion
- Write a paragraph that briefly summarizes and gives a scientific conclusion for the work that you did for part 1 and 2 this week.
Links
- User Page: Brandon Klein
- Team Page: The Class Whoopers
Assignments Pages
- Week 1 Assignment
- Week 2 Assignment
- Week 3 Assignment
- Week 4 Assignment
- Week 5 Assignment
- Week 6 Assignment
- Week 7 Assignment
- Week 8 Assignment
- Week 9 Assignment
- Week 10 Assignment
- Week 11 Assignment
- Week 12 Assignment
- No Week 13 Assignment
- Week 14 Assignment
- Week 15 Assignment
Individual Journal Entries
- Week 1 Individual Journal
- Week 2 Individual Journal
- Week 3 Individual Journal
- Week 4 Individual Journal
- Week 5 Individual Journal
- Week 6 Individual Journal
- Week 7 Individual Journal
- Week 8 Individual Journal
- Week 9 Individual Journal
- Week 10 Individual Journal
- Week 11 Individual Journal
- Week 12 Individual Journal
- No Week 13 Journal
- Week 14 Individual Journal
- Week 15 Individual Journal
- Week 1 Class Journal
- Week 2 Class Journal
- Week 3 Class Journal
- Week 4 Class Journal
- Week 5 Class Journal
- Week 6 Class Journal
- Week 7 Class Journal
- Week 8 Class Journal
- Week 9 Class Journal
- Week 10 Team Journal
- Week 11 Team Journal
- Week 12 Team Journal
- No Week 13 Journal
- Week 14 Team Journal
- Week 15 Team Journal